SMA is a rare genetic disease that affects the part of the nervous
system that controls voluntary muscle movement.
Understand SMA
What is SMA1
SMA is characterised by a loss of important cells in the spinal
cord called motor neurons. Over time, the breakdown of these
neurons leads to a gradual decline in muscle size and strength.
While primarily a childhood condition, SMA can be diagnosed
in adolescents, and sometimes beyond 18 years of age.1
Share this section
Copied
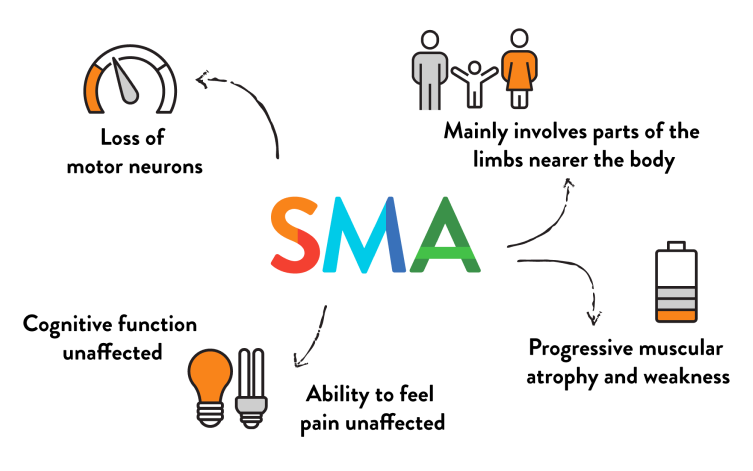
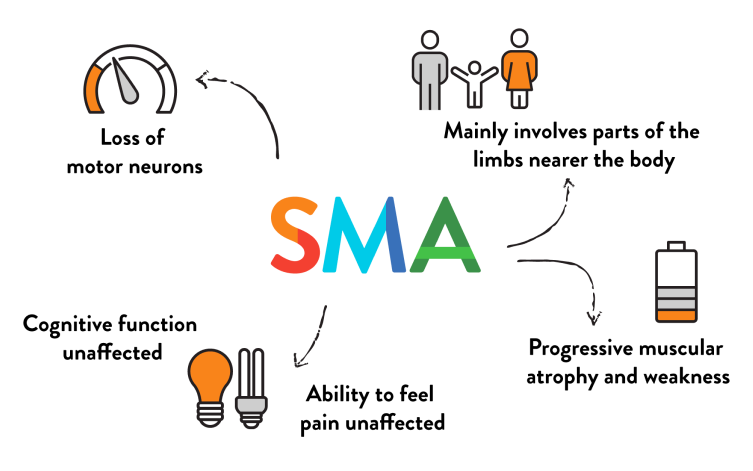
Key characteristics
Loss of motor neurons
Progressive muscular atrophy and muscle weakness
Muscles closer to the center of your body tends to be more affected than those further away but symptoms will vary according to disease severity
Can affect muscles used for feeding, swallowing, and breathing in some patients
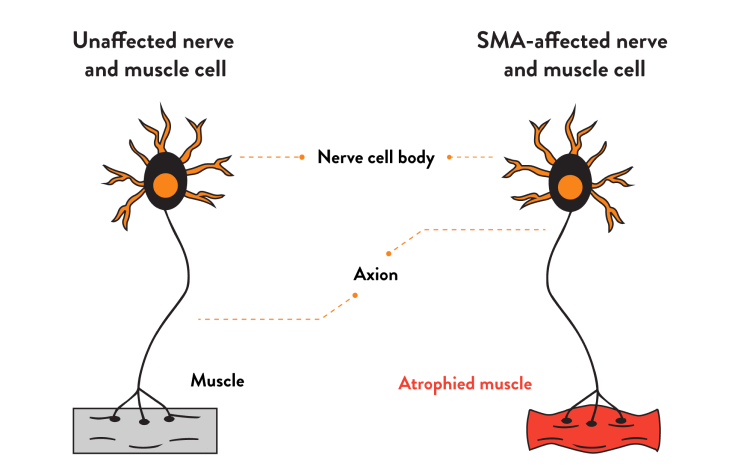
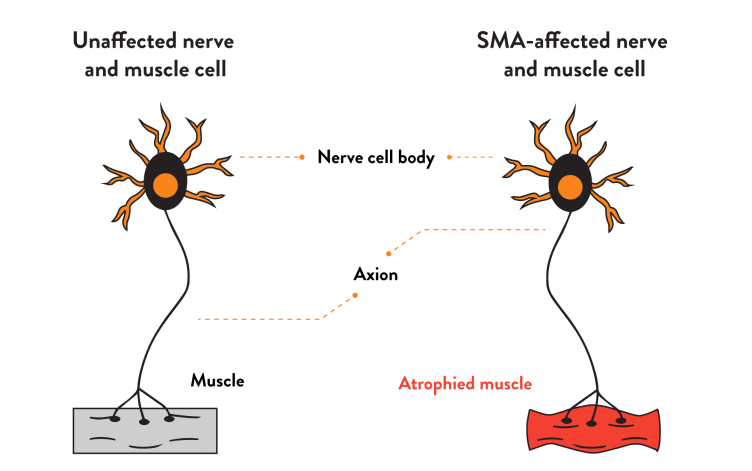
How are nerves affected in SMA?
Motor neurons are nerve cells that control muscle movement and strength by sending signals from the central nervous system (CNS) to muscle cells.
In SMA, as motor neurons deteriorate, muscles stop receiving signals from the CNS, and symptoms such as progressive muscle weakness and decreasing muscle mass (called atrophy) develop.
Understand SMA
What causes SMA?1
Share this section
Copied
SMN1 gene mutation
SMA is a genetic disorder caused by a mutation in the survival motor neuron 1 (SMN1) gene which prevents the production of the SMN protein necessary to maintain healthy motor neurons.
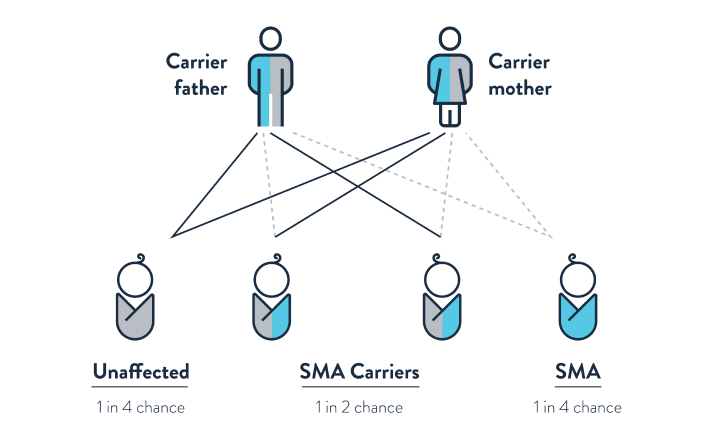
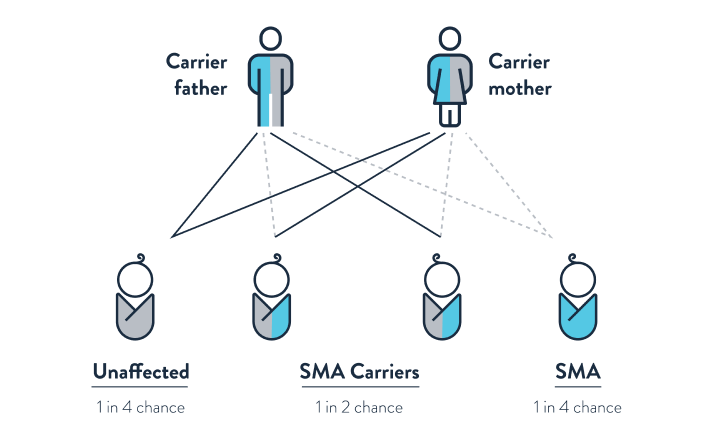
The role of genetics in SMA1
SMA is passed to a child when both parents are SMA carriers and exhibit no signs or symptoms. It is estimated that between 1 in 25 and 1 in 50 people are SMA carriers.
Understand SMA
Signs and symptoms1
SMA affects muscle strength and movement. Symptoms can vary greatly depending on age and severity of the condition.
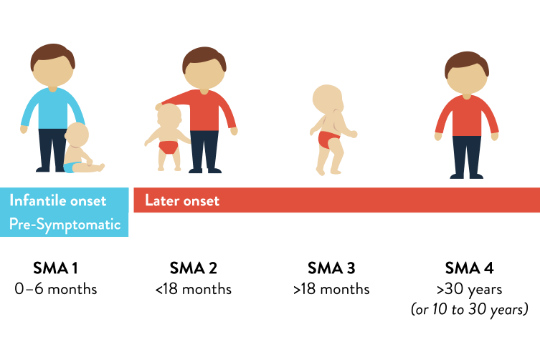
Pre-symptomatic SMA is the stage before muscle weakness appears in a person who is genetically diagnosed with SMA, and is typically identified with new born screening.
Symptomatic SMA often presents with weak muscles around the shoulders, thighs, and hips. Breathing and swallowing may also be impacted for some individuals.
Share this section
Copied
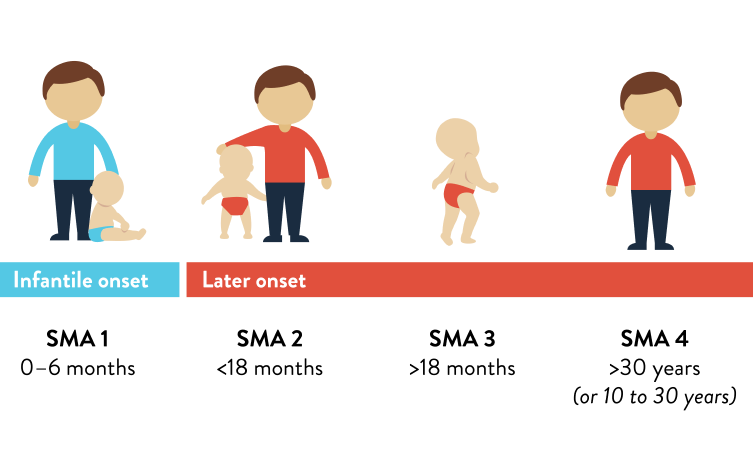
Are there different types of SMA?
SMA is a condition with a single spectrum and 5 different types, based on the age of symptom onset and the level of functional ability achieved. SMA types 1, 2 and 3 are most common, however, up to 25% of individuals cannot be accurately classified into a type. Type 0 occurs before birth.
Understand SMA
SMA Questions
Understanding SMA can mean asking a lot of questions. We have provided starter questions for information about SMA, and suggestions for questions you might like to ask your doctor to understand more about SMA.
Share this section
Copied
How rare is SMA?
In Australia, it is estimated that 7 in 1,000,000 people are living with SMA.1
1. Balaji L et al, Lancet Reg Health West Pac 2024. Nov 6: 53:101237
Is the SMN1 gene the only cause of SMA?
Approximately 95–98% of SMA diagnoses are caused by mutations to the SMN1 genes.1 This is known by neurologists and researchers as “5q SMA”. There are other, rarer, forms of SMA that may have similar symptoms. A neurologist will include these other conditions in their differential diagnosis.
The time until symptoms first appear can vary according to SMA type, but the genetic mutations that caused SMA are present before birth. SMA type 4 is an adult-onset form of SMA and symptom onset is generally around 30 years of age.1
1. Farrar MA et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355-368.
How do I know if I am an SMA carrier?
SMA carriers do not have signs and symptoms of SMA. The only way to determine SMA carrier status is with a genetic test.
It is thought that approximately 1 in 35 people are SMA carriers.1,2 However, individuals with a family history of SMA may have an increased risk of being an SMA carrier.
In making reproductive decisions, it may be helpful to consult with your physician to learn what mutation(s) are common in your family, and what appropriate tests may be required to detect these. Once your family mutation(s) are known, an appropriate test for your situation may be determined.
References:
1. Butchbach M. Front Mol Biosci 2016; 3: 7.
2. Kaczmarek A et al. Expert Opin Investig Drugs 2015; 24: 867–81.
Will our baby have SMA if my partner is an SMA carrier?
If both parents are SMA carriers, there is a 1 in 4 chance their child will have SMA. It may be helpful to consult your doctor about reproductive decisions and whether SMA carrier screening would be useful.1